Cleaning Validations and its Process in Pharmaceuticals:
Cleaning, its process & cleaning validation is a very important topics in the pharmaceutical industries. Even all professionals and organizations are always struggling to generate or understand the best cleaning processes and generations of CV documents.
First, we need to understand the meaning of cleanliness and its need in pharmaceuticals. Every pharmaceutical plant produces the drug with the help of equipment or vessels, so these manufacturing vessels & equipment need to be cleaned, to avoid contamination in the products.
Cleanness is the removal of organic or inorganic materials from objects or surfaces of the equipment which are in direct contact with the product while manufacturing the drug.
This is usually accomplished by cleansing with water, detergents, or disinfectants. Cleaning is required prior to disinfection or sanitization to remove organic and inorganic materials that may remain on surfaces and potentially interfere with the process’s effectiveness.
That’s why cleaning needs to be validated through the cleaning validation Process.

Cleaning validation is documented proof that a cleaning technique can reliably remove any products or cleaning agents that have been used on the equipment below the maximum permitted carry-over level that has been defined by science.
- A Lifecycle Approach

- Quality Risk Management
- Types of Residues
- Setting Acceptable Limits
- Cleaning Methods
- Sampling & Testing
- Matrix Approach
- Regulatory Framework
- Deficiencies
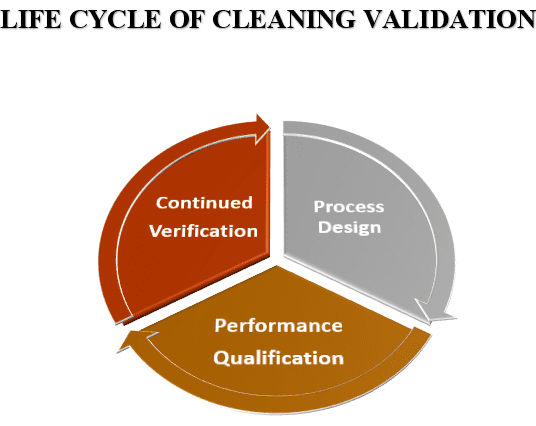
Implementing or understanding the cleaning validation program is very important for every pharma professional.
CV is an effective way to prevent product carry-over and cross-contamination so that the products are safe for patients.
In current practices, a risk-based approach is taken into account the throughout the life cycle of the cleaning process.
Periodic verification of sanitization effectiveness:
It should be carried out, with frequency based on risk assessment.
- The degree of contamination both before and after cleaning should be taken into account during validation.
- Product residues and perhaps disinfectant residues should be taken into consideration during chemical decontamination.
- Validating cleaning should be done frequently. Revalidation every year is advised.
- Limits should be set locally based on the investigation of both chemical and microbiological residues.
For better understanding, step-by-step procedure for validating the cleaning processes and maintaining the risk-based life cycle approach. While doing CV, a defined approach shall be
- Practically achievable
- Methodically verifiable
- Scientifically justifiable
In CV settable limits are set to support:
- Visual cleanliness (No visible residues on surfaces).
- Microbiological cleanliness (Absence of Bioburden & Endotoxin).
- Chemical cleanliness (Effective removal of APIs, Excipients, and Detergents).
Historically methods for cleaning validation:
- 10 ppm criterion.
- LD 50.
- 001 of dose method.
generated during the design stage of the cleaning process (trial batches, scale-up batches, and exhibit batches) provides insight into the efficiency of the designed process and confidence that cleaning is effective in removing drug residue from the product contact surface prior to cleaning validation.
Cleaning validation is written proof that a cleaning procedure used to remove residue from the equipment’s product contact surface meets established acceptance standards.
It is the process of gathering and analyzing data throughout the course of a product’s lifecycle to ascertain whether the cleaning method can consistently and reproducibly reduce the residue level to levels within the established acceptance standards.
For cleaning validation, which must be carried out during commercial batches, at least three successive runs should be taken into account.
It qualifies as a cleaning process performance standard. Following the completion of cleaning validation, routine and periodic cleaning verification approaches should be taken into consideration to guarantee that the cleaning process is in a validated state and state of control.
Through routine cleaning verification and sporadic cleaning verification, it can be kept up with and validated. On every batch of the product, routine cleaning verification can be carried out through visual inspection with minimal sampling for chemical analysis and at regular intervals for microbial examination.
In order to confirm that the current cleaning method is capable of satisfying the residue level within the established acceptance criteria, periodic cleaning verification is carried out on the worst-case scenario with one run.
Just simulating the cleaning validation run can be used to conduct chemical and microbiological samples.
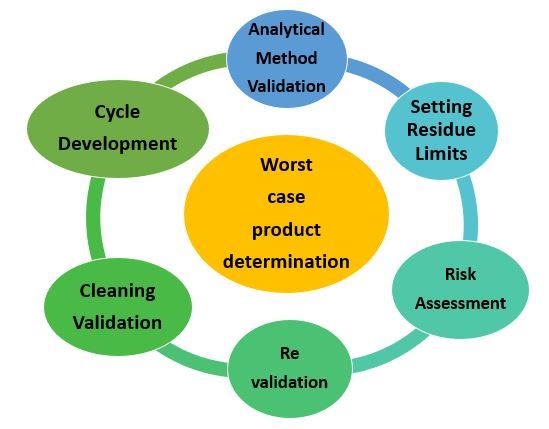
Risk-Based Approach for the CV Life Cycle:
- Cleaning Process Design.
- Cleaning Verification.
- Complete Qualification and analysis of the cleaning process.
- Verification of the cleaning process is in progress.
Cleaning Process Design Analysis:
The development of cleaning processes at a manufacturing facility or in research and development, then having those methods evaluated through cleaning, is the design of the cleaning process.
It covers the subsequent steps.
Cleanability Study and its Analysis:
To create a reliable and effective cleaning method, cleanability research should be carried out in a lab setting.
The factors that need to be taken into account are the nature of the product, the equipment’s maximum hold time before cleaning, the minimum cleaning processes parameters, such as the amount of time spent rinsing and flushing, the temperature of the water, and the chemical reactions, such as oxidation, reduction, hydrolysis, chelation, and reducing interfacial tension.
It needs to be used to remove microbiological and chemical residues. Each product must undergo a cleanability evaluation.
Types of Cleaning Validation :
Based on the following criteria:
- Identification & evaluation of potential residues
- Assigning appropriate acceptable residue limits.
Typical Residues:
- API (small molecule & macromolecule)
- Excipients
- Degradation Products
- Cleaning Agents
- Microbiological agents (Bioburden & Endotoxins)
Testing Methods:
In general, specialized procedures, i.e., methods where the analyte traces (ANALYTICAL METHODS) may be correctly recognized and quantified up to LOQ level utilizing instruments like HPLC, UPLC, GC, etc., will be given preference for evaluating the analyte.
The next step is non-specific approaches, which use methods like TOC, pH, Conductivity, Titration, etc. to evaluate the analyte for a group of substances collectively.
Specific or non-specific test procedures should be chosen and confirmed before usage depending on the type of analytes.
Cleaning Analytical Method:
Validation Prior to analyzing the analyte, the cleaning analytical procedure needs to be confirmed. Some of the criteria to be assessed during method validation include linearity, accuracy, precision, intermediate precision, recovery on all types of product contact parts, and hold time of the swab/rinse sample, among others.
Creation and classification of cleaning techniques:
The term “cleaning process” refers to the procedure for cleaning equipment, which requires the use of various cleaning process parameters.
The development of a cleaning procedure must resemble laboratory cleanability research while taking into account the following factors:
- Time—the length of time spent rinsing, flushing, and soaking.
- Action, including chemical and physical action. Scrubbing, soaking, flow rate, and pressure of PW/WFI are all examples of physical activity, while oxidation, reduction, saponification, reduction of interfacial tension, hydrolysis, chelation, etc. are some examples of chemical reaction.
- The temperature of the cleaning agent, PW, or WFI.
- The type of water or cleaning agent concentration utilized.
- Cleaning processes are separated into CIP and COP based on the location of the cleaning.
Based on the types of equipment :
- Manual vs. Automatic Process (Automation type).
- Equipment structure allows cleaning (CIP).
- Disassembly is required to clean out of place (COP).
The critical cleaning parameters :
- Selection of the detergent type and concentration.
- Pressure and temperature of the cleaning solution.
- cleaning process.
- Contact and rinse times with the detergent.
- The number of cleaning cycles there.
a) Clean-in-Place (CIP)
Equipment when cleaned in the place where equipment is located/ fixed either by using a CIP skid or by changing some piece of equipment, to perform cleaning.
b) Clean-out-of-Place (COP)
It is the process of cleaning when equipment parts are disassembled and transferred to the equipment wash area for cleaning.
Based on the nature of cleaning, the cleaning processes are divided into three parts: Automated, Semi-automated, and Manual.
Cleaning agent selection:
The cleaning agent is the substance that is used to enhance the cleaning process. During the selection of a cleaning agent, the following parameters must be considered
- It should be non-toxic
- It should be non-reactive with product contact MOC
- It should not produce excessive foam
During cleaning
- It should be removed easily from the equipment surface
- The composition of the cleaning agent should be known. As part of cleaning validation, the cleaning agent traces must be evaluated after cleaning of equipment. Specific or non-specific analytical methods can be considered for evaluation.
Cleaning Verification:
Considering the above-mentioned cleaning process parameters, a robust cleaning process should be designed.
During the design, the developed cleanability study must be considered and the process parameters must be replicated to achieve good cleanliness with careful consideration of the design and shape of the equipment.
Automated cleaning:
It is the process of cleaning where equipment or its parts is not disassembled and not moved to any other location for cleaning. The entire process happens without the intervention of the operator.
- Semi-automated cleaning: It is the process of cleaning where a few parts will be cleaned manually and some portions of equipment will be cleaned automatically. Semi-automated cleaning will happen with the intervention of the operator.
- Manual: It is the process of cleaning where equipment will be cleaned manually with the intervention of an operator.
Cleaning agent selection:
A cleaning agent is a substance that is used to enhance the cleaning process. During the selection of a cleaning agent, the following parameters must be considered
- It should be non-toxic
- It should be non-reactive with product contact MOC
- It should not produce excessive foam during cleaning
- It should be removed easily from the equipment surface
- The composition of the cleaning agent should be known.
As part of cleaning validation, the cleaning agent traces must be evaluated after cleaning of equipment. Specific or non-specific analytical methods can be considered for evaluation.
Cleaning Verification:
Considering the above-mentioned cleaning process parameters, a robust cleaning process should be designed.
During the design, the developed cleanability study must be considered and the process parameters must be replicated to achieve good cleanliness with careful consideration of the design and shape of the equipment.
Sampling Recovery:
Recoveries from all the product contact MOCs which is greater than 2% of the total equipment surface area should be determined.
The Recovery study should always be targeted to achieve not less than 70%. Correction factors must be considered while calculating the results.
Sampling:
Sampling shall be representative of the whole process. Samplings are of three types namely Swab sampling, Rinse sampling and Placebo sampling.
- Selection of equipment parts for sampling in a piece of equipment.
- Swab sampling/Physical sampling.
- Rinse sampling.
- Placebo sampling.
Conclusion:
When designing smart cleaning practices, properly defining and monitoring your cleaning goals is all that matters, taking into account both the complexity of cleaning and the significance of regulatory requirements. The choice of the analytical method and its validation give equal weight to the cleaning methods.
Firms should compile all the observations made during cleaning validation after the activities are successfully completed. To show that the predetermined aims are achieved and summarised, a report should be written. To demonstrate how they are closed, deviations and NCs should be outlined.
Look into your cleaning practices to see if there are any areas for consistency and improvement.
The operations should be accurately recorded in the document, with a clear “Pass/Fail” designation for your cleaning validation activities.
FAQ:
What is cleaning validation and its importance in the pharmaceutical industry?
Answer: Cleaning validation is an assurance there are no residuals or contamination of the previous product in the next manufacturing batch. This is documented proof that cleaning procedures for manufacturing equipment prevent product contamination is known as cleaning validation.
What is the cleaning validation process?
Answer: Clean-in-place Process: Usually a wash tank, recirculation pump, and related piping are used with fixed or revolving spray devices.
Clean-out-of-place: Frequently employed technique for automated parts washing through the cabinet or tunnel washers with cleansing, rinsing, and drying cycles.
Immersion Method: either mechanically stirring a cleaning agent in a process vessel to provide a cleaning effect, or simply soaking the process vessel in the cleaning agent.
Ultrasonic washing uses a tank with ultrasonic transducers to create cavitation, making it often more effective for delicate pieces like filling needles.
High-pressure Spraying: This technique uses continuously directed, high-pressure water or cleaning solution to remove any residues from the surface.
Manual Cleaning Method: The three most popular manual cleaning methods are wiping, sink brushing, and equipment brushing.
What is the significance of cleaning validation?
Answer: Cleaning validation’s primary goal is to demonstrate the efficiency and consistency of cleaning in a specific piece of pharmaceutical production machinery in order to prevent cross-contamination and adulteration of drug products with other active ingredients, such as unintended compounds or microbiological contamination, which can lead to a number of serious issues. Cleaning validation is also helpful in related studies, such as packaging component cleaning validation.
How to perform cleaning validation in the pharmaceutical industry?
Answer: Follow all the steps in the above articles, and take the help of the subject matter expert too.
What is 10 ppm criteria in cleaning validation?
Answer: In order to reduce cross-contamination in the product, the 10-ppm criterion for the allowable concentration of possible API in cleaning validation has been used. The 10-ppm threshold was created based on analytical constraints and acceptability estimations. A risk-based strategy should be universally adopted. this criteria involves a potency and toxicological study. Therefore, the allowable limits specified for cleaning shared equipment should be decided using the same thorough methodology based on health hazards.
For more information about Cleaning Validation and any draft documents, please write us at: admin@flairpharma.com
2 thoughts on “The prestige of Cleaning Validations and its Process in Pharmaceuticals”