The Dissolution Apparatus Principle includes the test conditions, dissolution medium, dosage form placement, sampling, sample analysis, data analysis, reporting, and documentation. DT is a vital tool used in the pharmaceutical industry to measure the rate at which a drug dissolves in a solution. This article explains the principles behind the dissolution apparatus and how it works to ensure the effectiveness of medications. Tablet dissolution is a process that measures the rate at which a solid dosage form, such as a tablet, disintegrates and releases its active pharmaceutical ingredient (API) into a surrounding liquid medium, typically water, under specified conditions. It is a critical quality control test performed during the formulation and manufacturing of pharmaceutical tablets to ensure that they meet their intended performance characteristics and that the API is released in a consistent and reproducible manner, this is the most important instrument in the quality control Lab.
Tablet dissolution testing is typically conducted using a dissolution apparatus, which consists of a set of vessels or cells that hold the tablets and a mechanism to agitate the liquid medium. The tablets are placed in the dissolution medium, and the dissolution apparatus is set to specific conditions, such as temperature, agitation speed, and pH, as per regulatory guidelines and product-specific requirements. As the test proceeds, samples of the dissolution medium are collected at predetermined time intervals, and the amount of API released from the tablets into the medium is measured using analytical techniques, such as spectrophotometry or chromatography.
The results of tablet dissolution testing provide important information on the dissolution profile of the tablets, including the rate and extent of API release, which can impact the bioavailability, efficacy, and safety of the pharmaceutical product. Dissolution testing is used during formulation development, process optimization, and batch release testing to ensure that tablets meet regulatory requirements and are of consistent quality.
Tablet Dissolution Testing
Tablet dissolution testing is a critical quality control test performed during the development, optimization, and manufacturing of pharmaceutical tablets to assess the rate and extent of drug dissolution from the tablets. It is an important parameter to ensure that the tablets release the active pharmaceutical ingredient (API) in a consistent and reproducible manner, which can impact the safety and efficacy of the final product.
The tablet dissolution testing is typically conducted using a dissolution apparatus, which consists of a set of vessels or cells that hold the tablets and a mechanism to agitate the dissolution medium, usually water or a buffer solution. The dissolution apparatus is set to specific conditions, such as temperature, agitation speed, and pH, as per regulatory guidelines and product-specific requirements.
Steps Involved in the dissolution testing & Dissolution Apparatus Principle
- Sample preparation: Tablets are carefully selected from the batch being tested, and any coating or film on the tablets is removed if necessary. The tablets are then accurately weighed or otherwise measured to ensure proper dosing.
- Dissolution medium preparation: The dissolution medium, typically water or a buffer solution, is prepared according to the specified conditions, such as temperature, pH, and composition, as per regulatory requirements or product-specific guidelines.
- Tablet placement: The tablets are carefully placed in the dissolution apparatus, usually in individual vessels or cells, ensuring that they are fully immersed in the dissolution medium without overlapping or touching.
- Dissolution testing: The dissolution apparatus is set to the specified conditions, and the dissolution test is initiated. The dissolution medium is agitated to maintain uniform conditions throughout the test. Samples of the dissolution medium are collected at predetermined time intervals.
- Sample analysis: The collected dissolution samples are analyzed using appropriate analytical techniques, such as spectrophotometry, chromatography, or other methods, to measure the concentration of the API released from the tablets into the dissolution medium.
- Data analysis: The dissolution data is analyzed to generate dissolution profiles, which show the rate and extent of drug dissolution from the tablets over time. The dissolution profiles are compared against predetermined acceptance criteria or regulatory requirements to assess the performance of the tablets.
- Reporting and documentation: The results of the tablet dissolution testing are documented in a report, including the dissolution profiles, sample analysis data, and any deviations from the acceptance criteria. The dissolution testing data is an important part of the batch release documentation and regulatory submissions.
Tablet dissolution testing is an important quality control test to ensure the consistency and quality of pharmaceutical tablets, and it is performed as per regulatory guidelines and product-specific requirements to assess the drug release characteristics and ensure the safety and efficacy of the final product.
The Working Principle of the dissolution test
The principal function of a dissolution tester, also known as a dissolution apparatus or dissolution testing equipment, is to measure the rate and extent of drug dissolution from solid dosage forms, such as tablets or capsules, in a liquid medium under specified conditions. The dissolution tester is used as a critical quality control tool during the development, optimization, and manufacturing of pharmaceutical tablets to ensure that they release the active pharmaceutical ingredient (API) in a consistent and reproducible manner.
The dissolution tester typically consists of several vessels or cells that hold the tablets or capsules and a mechanism to agitate the liquid medium, usually water or a buffer solution, to maintain uniform conditions during the dissolution test. The dissolution tester is designed to mimic the physiological conditions in the gastrointestinal tract, where the tablets or capsules are intended to release the API after administration. The dissolution apparatus is typically set to specific conditions, such as temperature, agitation speed, and pH, as per regulatory guidelines and product-specific requirements.
Main Functions of the dissolution tester:
- Standardized testing conditions: The dissolution tester provides controlled and standardized conditions, such as temperature, agitation speed, and pH, to ensure that the dissolution test is performed consistently and reproducibly, as required by regulatory guidelines.
- API release measurement: The dissolution tester allows for the measurement of the rate and extent of API release from the tablets or capsules into the dissolution medium over time. Samples of the dissolution medium are collected at predetermined time intervals during the dissolution test, and the concentration of the API in the samples is analyzed using appropriate analytical techniques.
- Dissolution profile generation: The dissolution tester helps generate dissolution profiles, which are graphical representations of the rate and extent of drug dissolution from the tablets or capsules over time. Dissolution profiles provide important information on the drug release characteristics, such as dissolution rate, dissolution efficiency, and dissolution behavior, which can impact the bioavailability, efficacy, and safety of the pharmaceutical product.
- Quality control testing: The dissolution tester is used as a quality control tool to assess the performance of tablets or capsules, ensuring that they meet predetermined acceptance criteria or regulatory requirements for drug release characteristics. The dissolution testing data is an important part of batch release documentation and regulatory submissions.
- Documentation and reporting: The dissolution tester facilitates the documentation and reporting of dissolution testing data, including the dissolution profiles, sample analysis data, and any deviations from the acceptance criteria. The dissolution testing data is documented in a report and used for quality assurance purposes.
Basic Principles of Dissolution Testing
Dissolution testing is a critical quality control test performed during the development, optimization, and manufacturing of pharmaceutical tablets, capsules, and other solid dosage forms to assess the rate and extent of drug dissolution. The basic principles of dissolution testing include the following:
- Test conditions: Dissolution testing is performed under controlled and standardized conditions, such as temperature, agitation speed, and pH, to ensure reproducibility and consistency of results. The test conditions are typically set according to regulatory guidelines and product-specific requirements, and they aim to mimic the physiological conditions in the gastrointestinal tract where the dosage form is intended to release the drug.
- Dissolution medium: A suitable dissolution medium, typically water or a buffer solution, is used to simulate the physiological environment in which the dosage form is intended to release the drug. The dissolution medium should be carefully prepared and maintained to meet the specified test conditions, and it should not interfere with the drug dissolution process.
- Dosage form placement: The dosage form, such as a tablet or capsule, is carefully placed in the dissolution apparatus, usually in individual vessels or cells, ensuring that it is fully immersed in the dissolution medium without overlapping or touching. The dosage form is typically positioned in a way that allows for adequate exposure of the drug to the dissolution medium to facilitate drug release.
- Sampling: Samples of the dissolution medium are collected at predetermined time intervals during the dissolution test. The sampling time points are usually selected to capture the critical stages of drug dissolution, such as the initial rapid release and the subsequent slower release phases. Care should be taken to ensure accurate and representative sampling to obtain reliable results.
- Sample analysis: The collected dissolution samples are analyzed using appropriate analytical techniques, such as spectrophotometry, chromatography, or other methods, to measure the concentration of the drug released from the dosage form into the dissolution medium. The analytical method used should be validated and reliable to ensure accurate and precise measurements.
- Data analysis: The dissolution data is analyzed to generate dissolution profiles, which show the rate and extent of drug dissolution from the dosage form over time. Dissolution profiles are graphical representations of the percentage of drug dissolved versus time and are used to assess the drug releases characteristics, such as dissolution rate, dissolution efficiency, and dissolution behavior. The dissolution profiles are compared against predetermined acceptance criteria or regulatory requirements to assess the performance of the dosage form.
- Reporting and documentation: The results of the dissolution testing are documented in a report, including the dissolution profiles, sample analysis data, and any deviations from the acceptance criteria. The dissolution testing data is an important part of the batch release documentation and regulatory submissions.
New Tools development in dissolution apparatus:
Dissolution apparatus, also known as dissolution testers or dissolution testing equipment, are standardized instruments used in pharmaceutical laboratories to perform dissolution testing on solid dosage forms, such as tablets and capsules. There are several types of dissolution apparatus that are commonly used, each with its own design and specifications, which are defined by regulatory agencies, such as the United States Pharmacopeia (USP) and the European Pharmacopoeia (Ph. Eur.). The most commonly used dissolution apparatus are:
- USP Apparatus 1 (Basket apparatus): This apparatus consists of a cylindrical basket that holds the dosage form and is suspended from a motor-driven spindle. The basket is typically made of stainless steel or other inert material, and it has a mesh or perforated bottom to allow for proper drug dissolution. This apparatus is commonly used for immediate-release dosage forms.
- USP Apparatus 2 (Paddle apparatus): This apparatus consists of a flat paddle that rotates in the dissolution medium and maintains a horizontal position during the test. The dosage form is typically placed on the bottom of the vessel and the paddle provides agitation to ensure proper drug dissolution. This apparatus is commonly used for both immediate-release and modified-release dosage forms.
- USP Apparatus 3 (Reciprocating cylinder apparatus): This apparatus consists of a cylindrical vessel in which a reciprocating cylinder moves up and down. The dosage form is placed inside the cylinder, and the reciprocating motion provides agitation to facilitate drug dissolution. This apparatus is commonly used for modified-release dosage forms.
- USP Apparatus 4 (Flow-through cell apparatus): This apparatus consists of a flow-through cell with two compartments separated by a porous membrane. The dissolution medium is continuously circulated through the cell, and the dosage form is placed in one compartment while the dissolution medium flows through the other compartment, allowing for drug dissolution. This apparatus is commonly used for extended-release dosage forms.
It is important to note that any new tools or modifications to dissolution apparatus should be thoroughly validated and properly documented to demonstrate their suitability for dissolution testing and to comply with regulatory requirements. Proper validation and documentation of the dissolution apparatus and any modifications are critical to ensuring the reliability and reproducibility of dissolution testing results, which are essential for evaluating the drug release characteristics and ensuring the quality of pharmaceutical dosage forms.
Common harmonized techniques used in dissolution apparatus
Harmonized techniques in dissolution apparatus refer to standardized methods that are accepted by multiple regulatory agencies, such as the United States Pharmacopeia (USP), the European Pharmacopoeia (Ph. Eur.), and other regulatory bodies. Harmonized techniques ensure consistency and comparability of dissolution test results across different laboratories and locations.
One example of a harmonized technique in dissolution apparatus is using standardized dissolution media, such as the USP or Ph. Eur. dissolution media. These media have predefined compositions and pH values and are commonly used in dissolution testing of pharmaceutical dosage forms. The use of standardized dissolution media helps to ensure consistent and reproducible results, as it minimizes variability in the dissolution medium composition, pH, and other critical parameters.
It is important for pharmaceutical manufacturers and laboratories to strictly adhere to harmonized techniques in dissolution apparatus to ensure the reliability, reproducibility, and accuracy of dissolution test results. This is essential for evaluating the drug release characteristics, ensuring the quality of pharmaceutical dosage forms, and complying with regulatory requirements.
Different Types of dissolution test apparatus
There are several types of dissolution test apparatus commonly used in pharmaceutical laboratories for testing the dissolution of solid dosage forms, such as tablets and capsules. These apparatuses are defined by regulatory agencies, such as the United States Pharmacopeia (USP) and the European Pharmacopoeia (Ph. Eur.), and are designed to provide standardized methods for evaluating drug release characteristics from dosage forms. Some of the common types of dissolution test apparatus are:
- Basket apparatus (USP Apparatus 1): This apparatus consists of a cylindrical basket made of stainless steel or other inert material that holds the dosage form and is suspended from a motor-driven spindle. The basket has a mesh or perforated bottom to allow for proper drug dissolution. It is commonly used for immediate-release dosage forms.
- Paddle apparatus (USP Apparatus 2): This apparatus consists of a flat paddle that rotates in the dissolution medium and maintains a horizontal position during the test. The dosage form is typically placed on the bottom of the vessel, and the paddle provides agitation to ensure proper drug dissolution. It is commonly used for both immediate-release and modified-release dosage forms.
- Reciprocating cylinder apparatus (USP Apparatus 3): This apparatus consists of a cylindrical vessel in which a reciprocating cylinder moves up and down. The dosage form is placed inside the cylinder, and the reciprocating motion provides agitation to facilitate drug dissolution. It is commonly used for modified-release dosage forms.
- Flow-through cell apparatus (USP Apparatus 4): This apparatus consists of a flow-through cell with two compartments separated by a porous membrane. The dissolution medium is continuously circulated through the cell, and the dosage form is placed in one compartment while the dissolution medium flows through the other compartment, allowing for drug dissolution. It is commonly used for extended-release dosage forms.
- Paddle over disk apparatus (Ph. Eur. Apparatus 2): This apparatus consists of a flat disk with a central hole and a paddle that rotates over the disk. The dosage form is placed on the disk, and the paddle provides agitation to facilitate drug dissolution. It is commonly used in European countries and is similar to the paddle apparatus (USP Apparatus 2).
- Reciprocating holder apparatus (Ph. Eur. Apparatus 3): This apparatus consists of a holder that holds the dosage form and moves up and down in the dissolution medium. The reciprocating motion provides agitation to facilitate drug dissolution. It is commonly used in European countries and is similar to the reciprocating cylinder apparatus (USP Apparatus 3).
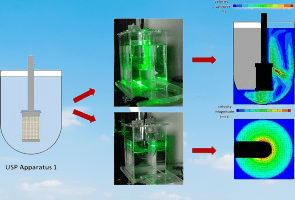
What are dissolution apparatuses and their uses?
Dissolution apparatus is a specialized piece of equipment used in pharmaceutical quality control (QC) laboratories to measure the rate and extent of drug dissolution from solid dosage forms, such as tablets, capsules, and powders, into a dissolution medium that simulates the physiological conditions in the gastrointestinal tract. The dissolution apparatus consists of a vessel, which holds the dissolution medium and the dosage form, and an agitation system, which agitates the medium to maintain uniform conditions throughout the test.
The main uses of dissolution apparatus are:
- Quality control: Dissolution testing is a critical quality control test performed to ensure that pharmaceutical dosage forms meet the required dissolution specifications, which are often defined in regulatory guidelines or pharmacopeial monographs.
- Drug development: Dissolution testing is used in the development of new pharmaceutical products to determine the optimal formulation, dosage form design, and manufacturing process parameters to achieve the desired drug release profile.
- Formulation optimization: Dissolution testing helps in optimizing the formulation of pharmaceutical products by evaluating the effect of various formulation factors, such as excipients, on drug dissolution rates.
- Batch-to-batch consistency: Dissolution testing is used to ensure batch-to-batch consistency in drug release performance of commercial pharmaceutical products, helping to maintain consistent product quality.
- Comparability studies: Dissolution testing is used in comparability studies to assess the similarity or equivalence of different batches or formulations of the same drug product, including generic drug products compared to reference-listed drug (RLD) products.
- Stability testing: Dissolution testing is used in stability studies to evaluate the stability of pharmaceutical products over time and under different storage conditions, providing information on the shelf-life of the product.
- Regulatory compliance: Dissolution testing is required by regulatory agencies worldwide as a part of the drug approval process to demonstrate the quality, safety, and efficacy of pharmaceutical products.
Overall, dissolution apparatus plays a crucial role in the pharmaceutical industry by providing important information about the performance and quality of pharmaceutical dosage forms, helping to ensure that.
The finding of Q value in dissolution
The Q value in dissolution refers to the cumulative amount of drug dissolved from a dosage form at a specific time during a dissolution test. It is often used as a parameter to evaluate the dissolution performance of a drug product.
The Q value is typically calculated by integrating the drug dissolution profile over time, usually up to a specific time point or time interval, using mathematical equations or software. The Q value represents the total amount of drug dissolved from the dosage form at that particular time, expressed as a percentage of the labeled amount of drug in the dosage form.
The Q value is used to assess the rate and extent of drug dissolution, and it is often compared to the dissolution specifications or acceptance criteria defined in regulatory guidelines or pharmacopeial monographs. It is used to evaluate the consistency of drug dissolution performance among different batches or formulations of the same drug product, as well as to assess the effect of formulation factors, manufacturing process parameters, or storage conditions on drug dissolution.
(Relative Standard Deviation)RSD limit for the dissolution test:
The RSD (Relative Standard Deviation) limit for dissolution testing is typically defined based on regulatory guidelines or pharmacopeial monographs, and it may vary depending on the specific drug product, dosage form, and dissolution medium being tested.
As per the United States Pharmacopeia (USP), the general acceptance criteria for RSD in dissolution testing are as follows:
- For a single-dose unit (e.g., tablet or capsule): RSD should be not more than 6% for the total drug released at each time point, and not more than 10% for the cumulative drug release at the end of the test.
- For a multiple-dose unit (e.g., extended-release product): RSD should be not more than 3% for the total drug released at each time point, and not more than 6% for the cumulative drug release at the end of the test.
It’s important to note that these are general guidelines, and specific RSD limits may vary depending on the drug product, regulatory requirements, and the specific dissolution method being used. Some drug products or dosage forms may have more stringent RSD limits based on their intended use, therapeutic indication, or regulatory requirements.
You may also read Alert & Action Limit Determination.
The dissolution rate calculation.
The dissolution rate is calculated by determining the amount of drug dissolved from a dosage form over a specified time period and expressing it as a rate, typically in units of mass or concentration per unit of time. The dissolution rate is an important parameter used to evaluate the performance of a drug product and its ability to release the drug in a controlled manner.
There are different methods to calculate the dissolution rate, depending on the units of measurement and the data available from the dissolution test. Here are two common methods:
- Mass-based dissolution rate: The mass-based dissolution rate is calculated by dividing the change in the mass of drug dissolved (Δm) from the dosage form by the time period (Δt) in which the dissolution occurred. The formula for calculating the mass-based dissolution rate is: Dissolution rate (mass) = Δm / Δt where Δm is the change in the mass of the drug dissolved and Δt is the time period in which the dissolution occurred. The dissolution rate is typically expressed in units of mass/time, such as mg/min or mg/hour.
- Concentration-based dissolution rate: The concentration-based dissolution rate is calculated by dividing the change in the concentration of the drug dissolved (ΔC) from the dosage form by the time period (Δt) in which the dissolution occurred. The formula for calculating the concentration-based dissolution rate is:
Dissolution rate (concentration) = ΔC / Δt
where ΔC is the change in the concentration of the drug dissolved and Δt is the time period in which the dissolution occurred.
The dissolution rate is typically expressed in units of concentration/time, such as mg/mL/min or %/hour.
Frequently Asked Question (FAQ’s)
What is the basic principle of dissolution?
Answer: The basic principle of dissolution is the process by which a solid substance (drug) dissolves in a solvent (dissolution medium) to form a solution. The rate at which a drug dissolves depends on factors such as the solubility of the drug, the surface area of the solid, the agitation or stirring of the medium, and the temperature of the dissolution medium.
How does a dissolution apparatus work?
Answer: A dissolution apparatus works by simulating the conditions in the gastrointestinal tract to measure the rate and extent of dissolution of a drug from its dosage form. The apparatus typically involves a vessel containing the dissolution medium, maintained at a specific temperature (usually 37°C), and an agitation mechanism (such as a paddle or basket) to ensure uniform mixing. The drug is placed in the medium, and samples are taken at specified intervals to measure the amount of drug dissolved.
What are the principal functions of dissolution testing?
Answer: The principal functions of dissolution testing are:
- Quality Control: To ensure batch-to-batch consistency of drug products.
- Formulation Development: To optimize the formulation and manufacturing process.
- Regulatory Approval: To demonstrate bioequivalence between a generic drug and the reference product.
- Product Compliance: To make product release decisions based on dissolution outcomes.
- Predicting In Vivo Performance: To estimate how the drug will behave in the human body.
What is the principle of USP Type 4 dissolution apparatus?
Answer: The USP Type 4 dissolution apparatus, also known as the Flow-Through Cell, operates on the principle of continuous flow. The dissolution medium is continuously pumped through a cell containing the drug product. This setup allows for the constant removal of dissolved drug, maintaining sink conditions and providing a more accurate simulation of in vivo conditions.
What is the main purpose of dissolution?
Answer: The main purpose of dissolution is to predict the in vivo drug release profile, ensure the consistency and quality of drug products, and to help in the development and optimization of drug formulations.
What is the concept of dissolution?
Answer: The concept of dissolution involves the process by which a solid drug dissolves in a liquid medium to form a solution. This process is influenced by various factors such as the physical and chemical properties of the drug, the composition and temperature of the medium, and the hydrodynamics within the dissolution vessel.
What are the 7 types of dissolution?
Answer: The common types of dissolution apparatus defined by USP are:
- USP Apparatus 1: Basket
- USP Apparatus 2: Paddle
- USP Apparatus 3: Reciprocating Cylinder
- USP Apparatus 4: Flow-Through Cell
- USP Apparatus 5: Paddle over Disk
- USP Apparatus 6: Cylinder
- USP Apparatus 7: Reciprocating Holder
What is the RSD limit for dissolution test?
Answer: The Relative Standard Deviation (RSD) limit for dissolution testing is typically set by regulatory guidelines and is usually around 10% or lower, depending on the specific requirements of the test and the dosage form being evaluated.
What is S1, S2, and S3 in dissolution?
Answer: S1, S2, and S3 are stages of dissolution testing:
- S1 Stage: Initial testing of 6 units; all units must meet the required specifications.
- S2 Stage: Additional 6 units tested if S1 fails; the average of 12 units must meet specifications, with no unit outside specific limits.
- S3 Stage: Further 12 units tested if S2 fails; the average of 24 units must meet specifications, with no more than 2 units outside specific limits.
Why is dissolution medium 900 ml?
Answer: The 900 ml volume of dissolution medium is used to ensure there is enough solvent to maintain sink conditions for the drug, preventing saturation and allowing for a more accurate assessment of the dissolution rate.
What is Q in dissolution?
Answer: Q is a parameter representing the quantity of drug dissolved at a specified time point, expressed as a percentage of the labeled amount. It is a critical value in determining if the drug product meets dissolution specifications.
What is f1 and f2 in dissolution?
Answer: f1 and f2 are similarity factors used to compare the dissolution profiles of two drug products:
- f1 (Difference Factor): Measures the percent difference between two profiles.
- f2 (Similarity Factor): Measures the closeness between two profiles, with values between 50-100 indicating similarity.
What is the principle of dissolution?
Answer: The principle of dissolution involves the process by which a solid substance dissolves in a liquid medium to form a solution. It is governed by the Noyes-Whitney equation, which relates the dissolution rate to the surface area of the solid, the solubility of the drug, and the concentration gradient in the medium.
What is disintegration time?
Answer: Disintegration time is the time required for a tablet or other solid dosage form to break down into smaller particles or granules. It is an important parameter for ensuring the drug is available for dissolution and subsequent absorption in the body.
How to test dissolution rate?
Answer: The dissolution rate is tested by placing the drug product in a dissolution apparatus, adding the dissolution medium, and agitating the solution. Samples are taken at specific time intervals and analyzed using techniques such as HPLC or UV-Vis spectroscopy to measure the amount of drug dissolved over time.
What are the three types of dissolution?
Answer: The three types of dissolution are:
- Intrinsic Dissolution: Dissolution of a pure substance under standard conditions.
- Apparent Dissolution: Dissolution of a formulated product, including excipients.
- Dissolution Testing: Routine testing of finished dosage forms to assess performance and quality.
What is the use of dissolution apparatus?
Answer: The dissolution apparatus is used to simulate in vivo conditions and measure the rate and extent of drug release from solid dosage forms. It is essential for quality control, formulation development, regulatory approval, and predicting in vivo performance.
What is the law of dissolution?
Answer: The law of dissolution is often referred to the Noyes-Whitney equation, which describes the dissolution rate of a solid in a liquid. It states that the dissolution rate is proportional to the surface area of the solid, the solubility of the drug, and the concentration gradient in the medium.
What is the rpm limit for dissolution?
Answer: The rpm (revolutions per minute) limit for dissolution testing typically ranges from 50 to 100 rpm, depending on the specific dissolution apparatus and method being used. For USP Apparatus 1 (Basket) and 2 (Paddle), common speeds are 50 or 75 rpm.
What is the mechanism of dissolution?
Answer: The mechanism of dissolution involves the breaking of intermolecular bonds in the solid phase, the diffusion of solute molecules into the surrounding solvent, and the establishment of a concentration gradient that drives further dissolution. It can be influenced by factors such as temperature, agitation, and the physicochemical properties of the drug.
What is USP Type 7 dissolution apparatus?
Answer: The USP Type 7 dissolution apparatus, also known as the Reciprocating Holder, is used for testing modified-release dosage forms. It consists of a reciprocating cylinder that moves vertically in and out of the dissolution medium, providing a more dynamic environment for assessing drug release.
Why 900 ml is used in dissolution test?
Answer: The 900 ml volume is used to ensure that there is sufficient dissolution medium to maintain sink conditions for most drugs. It prevents saturation of the solution, which could affect the accuracy and reproducibility of the dissolution test.
How to calculate dissolution?
Answer: Dissolution is calculated by measuring the amount of drug dissolved at specific time intervals, typically using HPLC or UV-Vis spectroscopy. The percentage of drug dissolved is then plotted against time to generate a dissolution profile.
What is USP apparatus 3?
Answer: USP Apparatus 3, also known as the Reciprocating Cylinder, is used for dissolution testing of extended-release dosage forms. It involves a reciprocating motion to simulate the dynamic conditions of the gastrointestinal tract.
What is dissolution method?
Answer: A dissolution method is a standardized procedure for testing the rate and extent of drug release from a dosage form. It includes details such as the type of dissolution apparatus, dissolution medium, temperature, agitation speed, sampling times, and analytical methods used to measure drug dissolution.
What is the dissolution rate?
Answer: The dissolution rate is the speed at which a drug dissolves in a dissolution medium. It is typically expressed as the amount of drug dissolved per unit time (e.g., mg/min).
What is the first step of dissolution?
Answer: The first step of dissolution is the disintegration of the solid dosage form, which increases the surface area exposed to the dissolution medium, allowing the drug to dissolve into the solution.
Great post! I was not aware of the importance of dissolution apparatus in the pharmaceutical industry. The principles you mentioned are very insightful. Can you please provide more information on the different types of dissolution apparatus and their applications?
Well explained blog post. The dissolution apparatus principle is indeed a crucial concept in pharmacy and this article has helped me understand it better. Thanks for sharing your expertise!
This post on the principles behind the dissolution apparatus is incredibly insightful! I appreciate how you broke down complex concepts into easily digestible sections. It’s great to see such up-to-date information for 2024. Looking forward to more posts like this!