Deviation in pharma refers to any departure from established standard operating procedures (SOPs) or specifications that could potentially impact the quality, safety, or efficacy of pharmaceutical products. Deviations can occur during any stage of the pharmaceutical product lifecycle, including manufacturing, testing, packaging, labeling, storage, distribution, or documentation. 
Deviation in Pharma
Deviations in the pharma industry are considered events or incidents that deviate from the expected or planned activities, processes, or outcomes. They can arise from a variety of reasons, such as equipment malfunction, human error, environmental factors, or unexpected events. Deviations can have different levels of severity, ranging from minor deviations that do not impact product quality, to major deviations that can lead to product recalls or regulatory non-compliance.
Planned and Unplanned Deviations in the pharma industry:
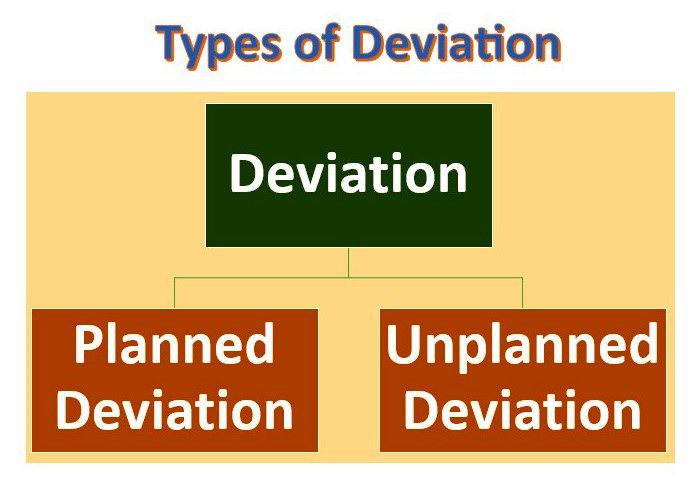
| Category | Planned Deviation | Unplanned Deviation |
| Definition | Deviations that are intentionally planned as part of a predefined process or procedure. | Deviations that occur unexpectedly or unintentionally during the course of normal operations. |
| Examples | Scheduled maintenance, process changes equipment upgrades and process upgrades. | Equipment malfunction, power outage, raw material quality issue, human error, contamination, environmental conditions, deviations from SOPs. |
| Purpose | To improve or optimize processes, comply with regulatory requirements, and implement changes based on quality risk assessments. | To investigate and correct unexpected or undesired events, identify root causes, prevent a recurrence, and ensure compliance with regulatory requirements. |
| Documentation | Planned change requests, change control documentation, SOP updates, validation protocols, and risk assessments. | The incident report, non-conformance report, investigation report, CAPA (Corrective and Preventive Action) report, and documentation updates. |
| Impact | Controlled impact on processes, potential for temporary disruption of operations. | Unpredictable impact on processes, the potential for significant disruption of operations, and potential for product quality issues. |
| Regulatory Requirements | May require notification or approval from regulatory agencies, and may impact product release or regulatory status. | May require immediate reporting to regulatory agencies, may require investigation and corrective action, and may impact product release or regulatory status. |
| Corrective Action | Predefined corrective action plans, and documented changes to procedures, training, or other quality controls. | The immediate investigation, root cause analysis, identification and implementation of corrective and preventive actions, documentation updates, and process improvements. |
Types of the deviations in pharma:
Deviations in the pharma industry, are often categorized into three levels of severity: critical, major, and minor. These categories are used to assess the potential impact of the deviation on product quality, safety, efficacy, and regulatory compliance. The classification of deviations as critical, major, or minor may vary slightly depending on the specific quality management system and standard operating procedures (SOPs) of a pharmaceutical company, but generally, the following definitions apply:
- Critical Deviation: A critical deviation in pharma is a serious non-conformance or failure that has a significant impact on product quality, safety, efficacy, or regulatory compliance. Critical deviations may result in the production of a non-conforming product that poses a significant risk to patient health or safety, or that does not comply with regulatory requirements. Critical deviations typically require immediate investigation, corrective actions, and regulatory notification to mitigate the risk and prevent a recurrence.
- Major Deviation: A major deviation in pharma is a non-conformance or failure that has a moderate impact on product quality, safety, efficacy, or regulatory compliance. Major deviations may result in the production of a product that does not fully meet the established quality standards or regulatory requirements, but that is not considered to pose an immediate or significant risk to patient health or safety. Major deviations typically require investigation, corrective actions, and documentation of the deviation and its resolution.
- Minor Deviation: A minor deviation is a non-conformance or failure that has a minimal impact on product quality, safety, efficacy, or regulatory compliance. Minor deviations may result in minor discrepancies or errors that do not significantly affect the quality or regulatory compliance of the product and are unlikely to pose any risk to patient health or safety. Minor deviations typically require investigation, documentation, and appropriate corrective actions, but may not require the same level of urgency or regulatory notification as critical or major deviations.
The classification of deviations as critical, major, or minor is important in determining the appropriate level of investigation, documentation, and corrective actions needed to address the deviation and prevent a recurrence. It helps prioritize deviations based on their severity and potential impact and ensures that appropriate resources and actions are allocated to address deviations in a timely and effective manner.

Deviation in Pharma
Deviation Management Process:
Deviations in the pharmaceutical industry are typically managed through a formal deviation management process, which includes the following key steps:
- Detection and reporting: Deviations are typically detected through routine monitoring, quality control activities, internal audits, or customer complaints. Once a deviation is identified, it needs to be promptly reported to the relevant personnel or departments responsible for managing the deviation process.
- Investigation: Deviations are investigated to determine their root causes and potential impact on product quality, safety, and efficacy. This may involve conducting a thorough analysis of the situation, gathering data and evidence, interviewing personnel involved, and performing scientific or technical assessments.
- Documentation and evaluation: Deviations are documented and evaluated to assess their severity, potential impact, and significance. This may include documenting the details of the deviation, performing risk assessments, and determining the appropriate corrective and preventive actions (CAPAs) to be taken.
- CAPA implementation: Based on the investigation and evaluation, appropriate CAPAs are identified and implemented to address the root causes of the deviation and prevent its recurrence. CAPAs may include corrective actions to address the immediate issue, as well as preventive actions to prevent similar deviations from occurring in the future.
- Verification and closure: The effectiveness of the implemented CAPAs is verified to ensure that the deviation has been adequately addressed. Once the CAPAs are verified and completed, the deviation is closed, and appropriate documentation is maintained for regulatory compliance and quality recordkeeping purposes.
Deviation management is a critical aspect of quality management in the pharmaceutical industry to ensure that products are manufactured, tested, and distributed according to established standards and regulations, and to prevent or mitigate potential risks to patient safety and product quality.
- The deviation is a departure from an approved instruction or established standard.
- The deviation is an unintended change.
- All deviations potentially impact on product’s fitness for purpose.
- All deviations potentially impact Regulatory Compliance.
Deviation in Pharma
Guidance for the deviation in pharma :
Any deviation from established procedures should be documented and explained. Critical deviations should be investigated and the investigation and its conclusions documented.
Procedures should exist for notifying responsible management in a timely manner of regulatory inspections of serious GMP deficiencies, product defects, quality-related complaints, and recalls. Regular quality reviews of APIs should be conducted such reviews should be annual and include at least:
- Failed batches
- Critical deviations
- Complaints/recalls
- Adequacy of corrective actions…
Deviations from approved standards of calibration on critical instruments should be investigated.
Incidents related to computerized systems that could affect the quality should be recorded and investigated.
Complete records should also be maintained for – Out-of-specification (OOS) investigations
Any deviation should be documented and explained. Any critical deviation should be investigated.
A validation report should be prepared … including recommending changes to correct deficiencies.
Types of Deviations in Pharma:
- Departures from established procedures (SOPs).
- Deviations during the manufacturing process (BPRs).
- Deviating materials.
- Aberrant results/yields.
- OOS / OOT / OOE.
- Out of Limits (Environmental Monitoring).
- Equipment malfunction.
- Unusual occurrences/observations.
Technical details about Deviation in Pharma.
- Equipment out of calibration tolerance.
- Equipment was found to be malfunctioning during routine maintenance.
- Equipment breakdown during production.
- Computer software malfunction.
- Power losses whilst in operation.
- Any alarm situation affecting critical processes (CIP/SIP, Production).
- Any persistent alarm affecting the building, and utilities.
Deviations/Incidents/Deficiencies (DIDs)
- SOPs are needed in order to define “DIDs”
- All “DIDs” have to be reported to the Quality Unit
- All “DIDs” must be monitored in order to ensure timely evaluation and corrective actions
- The whole organization must be alert to “DIDs”
- Better to over-report than to under-report
- DIDs are the lifeblood of the CAPA system
Steps of Deviation in Pharma:
- Describe clearly the observed deviation:
- What has happened/ what did one see?
- When did it happen: day, point of time, step in the process.
- How was it discovered?
- Who has it discovered?
- Where?
- What immediate corrective actions (if any) were taken?
Important Key Points of deviaton in pharma :
- It is all about establishing the facts, not finding the guilty
- Timeliness of reporting is very important (preferably within 24 hours after detection of deviation)
- Thorough root cause analysis
- Thorough impact assessment
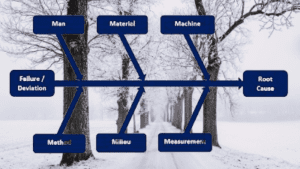
Deviation: Root cause analysis
- Root cause analysis needed to define appropriate actions
- Wrong root cause = wrong preventative actions
- Root cause analysis must be systematic
- Five M principle (Material, Method, Machine, Man, Mother Nature
- Ishikawa Diagram (Fishbone)
- Kepner Tregoe
- Why, Why, Why, Why
- Root cause analysis must be documented to show what was excluded as well as what was considered.
- Investigate in enough detail: 3 to 5 times the “WHY” question
- Essential in relation to:
- Impact assessment
- Preventative actions
Example of Deviation in pharma:
The temperature of the tray dryer exceeded the specification of 45 °C.
- Why (1): Controller set the temperature too high
- Why (2): The monitoring probe was not at the correct location
- Why (3): Monitoring probe misaligned during loading of equipment
- Why (4): Probe too close to product trays
Prevention:
- Relocate probe (technical solution)
- Check-in BPR after loading that probe is correctly positioned (Organisation solution)
Both solutions solve recognized Failure Mode.
ROOT CAUSE ANALYSIS: common failures
- Hunting the guilty
- “Jumping to conclusions”
- Making assumptions without verification
- Too many uncoordinated investigations (conflicting conclusions)
- Endless investigating
- Investigation not focused (scope too narrow, too wide)
- Stopping after finding the first possible root cause
- Focus on symptoms
- Immediately going into details
- Lack of proper SMEs
- Too late (weeks/months after the event)
- No real conclusions reached
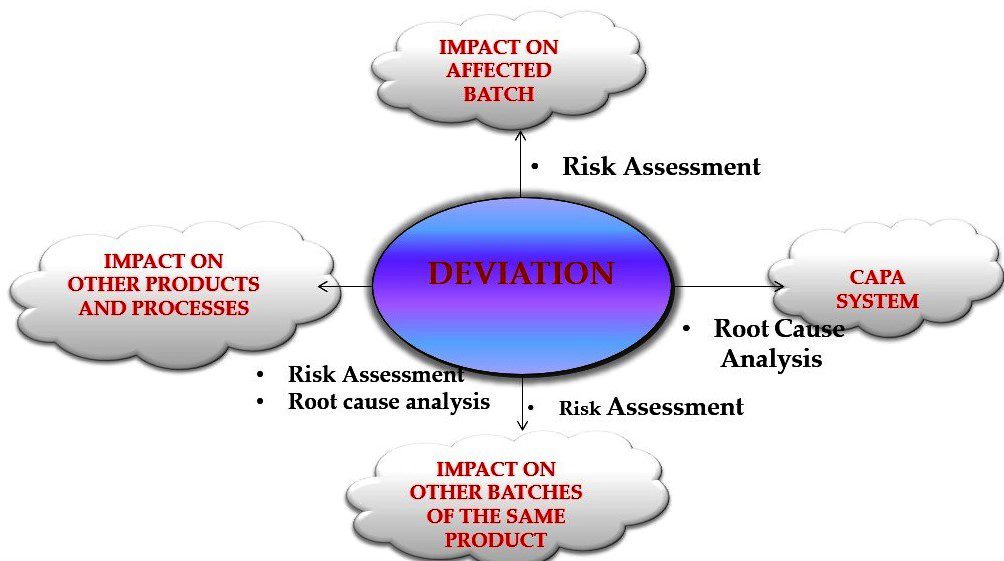
The key points related to the handling of deviations in the pharma industry:
- Prompt detection and reporting: Deviations should be promptly detected and reported through established procedures or systems, such as routine monitoring, quality control activities, internal audits, or customer complaints. Timely reporting allows for swift investigation and resolution of deviations.
- Thorough investigation: Deviations should be thoroughly investigated to determine their root causes and potential impact on product quality, safety, and efficacy. This may involve conducting a comprehensive analysis of the situation, gathering data and evidence, interviewing personnel involved, and performing scientific or technical assessments.
- Risk assessment: Risk assessment should be performed to evaluate the severity, potential impact, and significance of the deviation. This helps in prioritizing deviations and determining appropriate corrective and preventive actions (CAPAs) be taken.
- CAPA implementation: CAPAs should be identified and implemented based on the investigation and risk assessment findings. CAPAs may include corrective actions to address the immediate issue and preventive actions to prevent similar deviations from occurring in the future. CAPAs should be documented and their implementation progress tracked and monitored.
- Verification and closure: The effectiveness of the implemented CAPAs should be verified to ensure that the deviation has been adequately addressed. This may involve conducting follow-up inspections, testing, or other verification activities. Once the CAPAs are verified and completed, the deviation is closed, and appropriate documentation is maintained for regulatory compliance and quality recordkeeping purposes.
- Documentation and reporting: Throughout the deviation handling process, all relevant details, investigation findings, and CAPAs should be documented in a formal deviation report or electronic system. This documentation is used for regulatory reporting, quality recordkeeping, and future reference.
- Regulatory compliance: Deviation handling should comply with relevant regulatory requirements, such as Good Manufacturing Practices (GMP), Good Laboratory Practices (GLP), and other applicable regulations. It is important to ensure that all deviations are managed in accordance with regulatory guidelines and requirements.
- Continuous improvement: Deviations and their resolutions should be used as opportunities for continuous improvement. Lessons learned from deviations can be used to update procedures, training, and quality management systems to prevent similar deviations in the future.
- Cross-functional collaboration: Handling of deviations often requires cross-functional collaboration, involving personnel from different departments or functions within the pharmaceutical plant. Effective communication and collaboration among various stakeholders are crucial for the timely and effective resolution of deviations.
- Training and competency: Personnel involved in the deviation handling process should be adequately trained and competent in deviation management procedures, investigation techniques, and CAPA implementation. Regular training and competency assessments should be conducted to ensure that personnel is equipped with the necessary skills and knowledge to handle deviations effectively.
Overall, the key points of handling deviations in the pharmaceutical industry involve prompt detection, thorough investigation, risk assessment, CAPA implementation, verification and closure, documentation and reporting, regulatory compliance, continuous improvement, cross-functional collaboration, and personnel training and competency. Following these key points can help ensure effective management and resolution of deviations, and maintain product quality, safety, and efficacy in the pharmaceutical manufacturing process.
What Is the Difference Between Incident and Deviation?
| Aspect | Incident | Deviation |
| Definition | Unexpected events or occurrences that may impact product safety, quality, or efficacy | Departure from established procedures or specifications resulting in a non-conformance or failure of a process or product. |
| Documentation | May or may not require a formal report. | Requires a formal deviation report or non-conformance report (NCR). |
| Investigation | .May or may not require formal investigation | Requires formal investigation, root cause analysis, and documentation. |
| Severity | Generally broader in scope, may not always result in the issuance of a formal report. | Specific to a particular process or product, classified as critical, major, or minor. |
| Corrective and Preventive Actions | Actions taken or planned may vary based on the incident and its severity. | Formal corrective and preventive actions (CAPA) are typically required to address the non-conformance and prevent a recurrence. |
| Management Process | Managed through the incident reporting process, may or may not result in a formal deviation report. | Managed through a formal deviation handling process as part of the quality management system |
What Is a Deviation Report?
Answer: A deviation in pharma report, also known as a deviation or non-conformance report (NCR), is a formal document that is generated when a deviation or non-conformance is identified during the manufacturing, testing, packaging, labeling, storage, or distribution of pharmaceutical products. Deviation reports are an important part of the quality management system in the pharmaceutical industry and are used to capture, investigate, and document any departures from established procedures, Microbiology deviation specifications, or regulations.
A deviation in pharma report typically includes the following information:
- Deviation Details: This section provides a concise description of the nature of the deviation, including the date, time, location, and personnel involved. It may also include information such as the product or process affected, the deviation category (critical, major, minor), and the potential impact on product quality, safety, efficacy, or regulatory compliance.
- Investigation and Root Cause Analysis: This section outlines the investigation conducted to determine the root cause of the deviation. It may include details on the investigation methods used, data collected, and analysis performed to identify the underlying reasons for the deviation. Root cause analysis is an important step in understanding the underlying causes of the deviation and implementing effective corrective actions.
- Corrective and Preventive Actions (CAPA): This section outlines the corrective and preventive actions taken or planned to address the deviation and prevent a recurrence. It may include details on the specific actions taken, timelines for completion, responsible parties, and verification of effectiveness. CAPA is a critical aspect of deviation management, as it ensures that appropriate actions are taken to address the root cause of the deviation and prevent its recurrence.
- Documentation and Approval: This section includes the documentation of the deviation report, including signatures of the personnel involved in the investigation and approval of the corrective and preventive actions. It serves as a formal record of the deviation, investigation, and actions taken, and is typically maintained as part of the quality records in the pharmaceutical company.
- Follow-up and Closure: This section outlines the follow-up activities, including verification of the effectiveness of the corrective and preventive actions, and closure of the deviation report. It may include details on the verification activities performed, timelines, and documentation of the closure of the deviation report.
Deviation in pharma reports is an essential tool for managing and documenting deviations in the pharma industry, ensuring that deviations are properly investigated, addressed, and documented in accordance with regulatory requirements and good manufacturing practices (GMP).
2 thoughts on “Deviation in Pharma 2023”